
von
Eine bahnbrechende Studie, die Gesamtgenomdaten nutzt, hat unser Verständnis der Vogelevolution revolutioniert und neue Erkenntnisse über die evolutionären Beziehungen zwischen Vogelarten enthüllt. Durch die Analyse eines umfassenden Datensatzes von 363 Vogelarten hat die Forschung den Stammbaum der Vögel neu definiert, vier Hauptzweige innerhalb der neuen Vögel identifiziert und Licht auf die komplexe Evolutionsgeschichte der Vögel geworfen. Bildnachweis: SciTechDaily.com
Die bislang umfassendste Genomstudie hat gezeigt, wie sich Vögel nach einem Massensterben auf der ganzen Welt ausbreiteten.
Vögel sind heute die einzige überlebende Linie der Dinosaurier. Vor etwa 66 Millionen Jahren, beim Übergang von Kreidezeit Bis zum Paläogen (K-Pg-Grenze) führte ein katastrophales Aussterben zum Aussterben aller Nicht-Vogel-Dinosaurier, was den Vögeln die Möglichkeit bot, sich schnell zu diversifizieren und ein breites Spektrum ökologischer Nischen zu besetzen.
Neoaves, eine vielfältige Gruppe, die etwa 95 % aller Vögel umfasst Klassifizieren Heute ist er aus dieser Strahlung herausgekommen. Von hoch aufragenden Andenkondoren bis hin zu winzigen Kolibris, die durch tropische Wälder flattern, umfassen Newaves eine erstaunliche Vielfalt an Formen und Funktionen.
Trotz erheblicher Bemühungen, die Evolutionsgeschichte der Vögel und die Auswirkungen des K-Pg-Ereignisses mithilfe morphologischer und molekularer Daten zu rekonstruieren, sind die genaue Verzweigungsreihenfolge und die Beziehungen zwischen neuen Vogellinien weiterhin umstritten.
„Frühere Studien, die mit kleinen Datensätzen aus verschiedenen Genomregionen durchgeführt wurden, lieferten oft widersprüchliche Ergebnisse hinsichtlich der Topologie des Vogelbaums“, sagte Guojie Zhang, Hauptautorin des Papiers, Lehrstuhlinhaber für Evolutionsbiologie an der Zhejiang-Universität und einer der Initiatoren. B10K-Projekt. „In dieser Studie haben wir zum ersten Mal vollständige genomweite Daten verwendet, um einen Baum von Vogelarten aus fast allen repräsentativen Familien zu erstellen.“
Fortschritte bei Genomdaten
Dieser gesamte Genomdatensatz wurde vom B10K-Konsortium in seiner zweiten Phase erstellt und umfasst 363 Vogelarten, die alle wichtigen Vogellinien abdecken.
Der neue Stammbaum eröffnet neue Horizonte auf der langen Reise, die Geheimnisse der Vogelentwicklung aufzudecken. Laut diesem aktualisierten Vogelstammbaum gehörte eine Gruppe bestehend aus Flamingos und Krähen (Merandornithes genannt) zu den ersten neuen Vogellinien, die sich entwickelten.
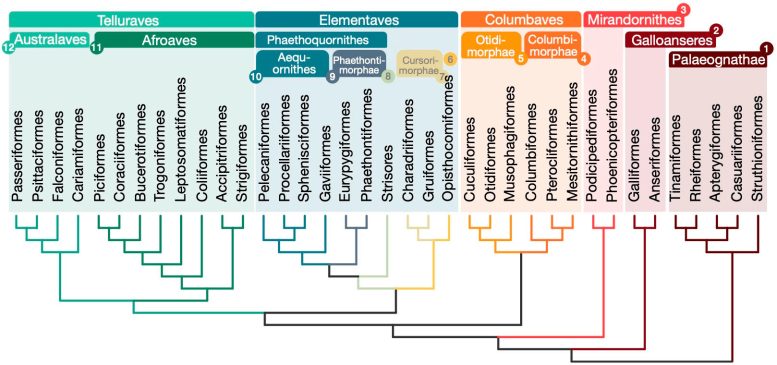
Beziehungen zwischen 363 Vogelarten basierend auf 63.430 intergenen Positionen. Bildnachweis: Josephine Stiller
Der neue Baum stellt die Organisation der Neoaven in Frage, indem er diese große Gruppe in vier Hauptzweige einteilt: Mirandornithes, Columbaves, Elementaves und Telluraves. Elementaves ist eine neu vorgeschlagene Sammlung, die ca. umfasst. 14 % aller modernen Vogelarten, darunter so unterschiedliche Gruppen wie der rätselhafte Hoatzin, Watvögel, Kolibris und tropische Vögel.
Die Namen „Elementaven“ spiegeln die bemerkenswerte Vielfalt der Umweltbereiche der Gruppe wider, die die Hauptelemente Erde, Luft und Wasser repräsentieren. Dieser neue Stammbaum löst einige langjährige Debatten über die Beziehungen zwischen Vogelarten und legt eine solide Grundlage für die Untersuchung der Vogelentwicklung und der Merkmalsentwicklung.
Auswirkungen einer umfassenden Genomanalyse
Unter Verwendung vollständiger Genomdaten von 363 Vogelarten ist dies der größte Datensatz, der jemals für evolutionäre Analysen von Vögeln verwendet wurde. Das Team baute eine neue Pipeline auf, um mehr als 150.000 über das Genom verteilte Regionen zu extrahieren.
„Wir haben evolutionäre Beziehungen im gesamten Genom charakterisiert und Muster identifiziert, die mit genomischen Kontext- und Sequenzeigenschaften verbunden sind“, sagte Josephine Stiller, Hauptautorin dieser Studie und Assistenzprofessorin für Evolutionsbiologie an der Universität Kopenhagen. Teile des Genoms, zum Beispiel einzelne Chromosomen oder proteinkodierende Gene, unterstützen oft sehr unterschiedliche Bäume. Dies erklärt wahrscheinlich, warum Studien, die nur bestimmte Teile des Genoms analysierten, widersprüchlich sind.
Die Zusammenstellung großer, qualitativ hochwertiger Datenmengen war für die Erstellung eines robusten phylogenetischen Stammbaums von entscheidender Bedeutung. Das Team stellte fest, dass für die meisten Branchen ein Konsens über ihre Beziehungen erzielt werden kann, wenn eine ausreichende Datenmenge verwendet wird. Aber die evolutionäre Position einiger Vogelgruppen wie Eulen und Falken bleibt selbst mit vollständigen Genomdaten rätselhaft.
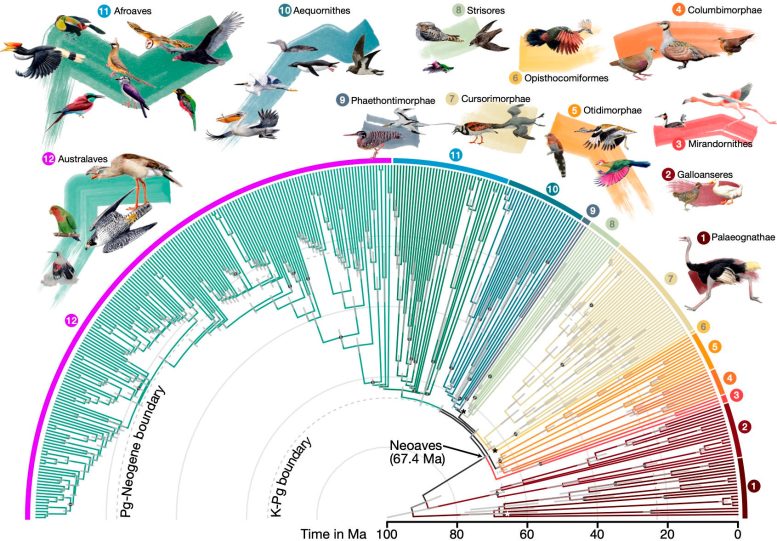
Divergenzzeiten für 363 Vogelarten basierend auf 63.430 intergenen Positionen. Bildnachweis: Josephine Stiller
„Mehr Daten führen nicht unbedingt zu einer besseren Lösung“, sagte Zhang. „Der Grund dafür kann eine komplexe Evolutionsgeschichte sein, wie zum Beispiel eine uralte Paarung zwischen zwei Abstammungslinien, eine unvollständige Abstammungssortierung, eine Anziehungskraft nach langen Zweigen und Voreingenommenheit.“ DNA Sequenzinhalte, die alle die Rekonstruktion phylogenetischer Bäume beeinträchtigen können.
Die Studie liefert neue Erkenntnisse darüber, welche dieser Faktoren die Äste des Baumes beeinflussen, und liefert so ein umfassenderes und wahrheitsgetreueres Bild der Herkunft dieser Vogelgruppen. Die Studie legt auch eine genauere Zeitskala für die Vielfalt moderner Vögel nahe, was darauf hindeutet, dass eine schnelle Strahlung an oder in der Nähe des Massenaussterbens an der Kreide-Paläogen-Grenze (K-Pg) und in geringerem Maße kurz nach dem Paläogen-Neogen auftrat . die Grenze.
Die Forscher fanden heraus, dass diese Strahlungen mit bemerkenswerten genetischen und morphologischen Veränderungen bei Vögeln einhergingen, darunter höhere Mutationsraten, kleinere Körpergrößen, größere Gehirne und größere effektive Populationsgrößen. „Dies zeigt die Leistungsfähigkeit der vergleichenden Genomik: Durch den Vergleich der Genome lebender Arten können wir Spuren von Ereignissen erkennen, die vor 66 Millionen Jahren stattgefunden haben“, sagte Steller.
„Unsere Arbeit hat viele traditionelle Ansichten über die Evolutionsgeschichte der Vögel verändert. Dieser neue Stammbaum wird als leistungsstarkes Rückgrat für die Kartierung der Evolutionsgeschichte aller Vogelarten mit wichtigen Implikationen für die ornithologische Forschung und Biodiversitätsstudien dienen“, schloss Zhang.
Referenz: „Die Komplexität der Vogelevolution, die durch Genome auf Familienebene aufgedeckt wird“ von Josephine Steller, Shaohong Feng, Abid Chowdhury, Iker Rivas Gonzalez, David A. Duchenne, Qi Fang, Yuan Deng, Alexei Kozlov, Alexandros Stamatakis, James Claramont, Jacqueline M. T. Nguyen, Simone Yu Hu, Brant C. Faircloth, Julia Haag, Peter Hood, Joel Cracraft, Metin Balaban, Owen May, Guangjie Chen, Rongxing Gao , Qingran Zhou, Yulong Xie, Zijian Huang, Chen Cao, Qi Yan, Hu A. Ogilvy, Louay Nakhleh, Bint Lindo, Benoit Morel, John Fieldsoe, Peter A. Hosner, Root R. da Fonseca, Bent Petersen, Joseph A. Tobias, Tamas Szekely, Jonathan David Kennedy, Andrew Hart Reeve, Andras Lecker, Martin Starvander, Agostino Antunes, Dieter Thomas Tietz, Mads Bertelsen, Fumin Li, Carsten Rahbek, Gary R. Graves, Mikael H. Scherup, Tandy Warno, Edward L . Braun, M. Thomas B. Gilbert, Eric D. Jarvis, Siavash Merab und Guji Chang, 1. April 2024, Natur.
doi: 10.1038/s41586-024-07323-1

„Studioso televisivo sottilmente affascinante. Organizzatore certificato. Imprenditore. Amichevole fanatico di Twitter. Fanatico della cultura pop. Appassionato di cibo.“